
Ob Umwelt- und Naturwissenschaften, Physik, Medizin, Ingenieurswesen oder Archäologie und Kunst: Sie alle brauchen für Simulationen, Berechnungen und Visualisierungen viel Rechenkraft und daher leistungsfähige Computer. Tauchen Sie mit unseren Berichten ein in die Welt von Wissenschaft und Forschung. Entdecken Sie neue Galaxien, das Innerste des Menschen, die Folgen von Klimawandel, historische Säle und Bauten, faszinierende Kunstwerke und mehr. Oder lesen Sie von neuesten Errungenschaften in der IT.

Eine internationale Forschungskooperation berechnete auf dem SuperMUC-NG des Leibniz-Rechenzentrums die bisher größten…

In ihrer Summer School für Supercomputing vermitteln das Leibniz-Rechenzentrum und das IT4Innovations National…

Neueste Technik für den nächsten Supercomputer des Leibniz-Rechenzentrums (LRZ): Er wird mit der Vera Rubin-Architektur…

Forschende der Ludwig-Maximilians-Universität München (LMU) suchen nach Strategien, RNA-basierte Medikamente…

Künstliche Neuronale Netze werden immer größer und komplexer – und brauchen daher fürs Training mit Daten immer mehr…

Das hat System im Gauss Center for Supercomputing: Die Leiter der drei zugehörigen Rechenzentren teilen sich die…

Beste Stimmung, bestes Wetter und jede Menge spannender Gespräche: Die Teilnahme des Leibniz-Rechenzentrums (LRZ) mit…
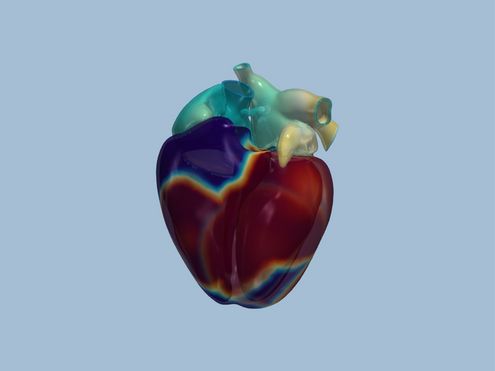
deal.II ist eine Programm-Bibliothek voller mathematischer Werkzeuge für die Berechnung von Strömungen oder für…

100 Arbeitstage hat das neue CoolMUC-System bald erfolgreich absolviert: Die ersten praktischen Erfahrungen mit dem…